Combining Data
Last updated on 2023-04-27 | Edit this page
Overview
Questions
- How can I combine dataframes?
- How do I handle missing or incomplete data mappings?
Objectives
- Determine which kind of combination is desired between two dataframes.
- Combine two dataframes row-wise or column-wise.
- Identify the different types of joins.
Key Points
- Concatenate dataframes to add additional rows.
- Merge/join data frames to add additional columns.
- Change the
onargument to choose what is matched between dataframes when joining. - The different types of joins control how missing data is handled for the left and right dataframes.
There are a variety of ways we might want to combine data when performing a data analysis. We can generally group these into concatenating (sometimes called appending) and merging (sometimes called joining).
We will continue to use the rnaseq dataset:
PYTHON
import pandas as pd
url = "https://raw.githubusercontent.com/ccb-hms/workbench-python-workshop/main/episodes/data/rnaseq.csv"
rnaseq_df = pd.read_csv(url, index_col=0)Concatenate dataframes to add additional rows.
When we want to combine two dataframes by adding one as additional rows, we concatenate them together. This if often the case if our observations are spread out over multiple files. To simulate this, let’s make two miniature versions of `rnaseq_df`` with the first and last 10 rows of the data:
PYTHON
rnaseq_mini = rnaseq_df.loc[:,["sample", "expression"]].head(10)
rnaseq_mini_tail = rnaseq_df.loc[:,["sample", "expression"]].tail(10)
print(rnaseq_mini)
print(rnaseq_mini_tail)OUTPUT
sample expression
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Klk6 GSM2545336 287
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
sample expression
gene
Dusp27 GSM2545380 15
Mael GSM2545380 4
Gm16418 GSM2545380 16
Gm16701 GSM2545380 181
Aldh9a1 GSM2545380 1770
Mgst3 GSM2545380 2151
Lrrc52 GSM2545380 5
Rxrg GSM2545380 49
Lmx1a GSM2545380 72
Pbx1 GSM2545380 4795We can then concatenate the dataframes using the pandas function
concat.
OUTPUT
sample expression
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Klk6 GSM2545336 287
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
Dusp27 GSM2545380 15
Mael GSM2545380 4
Gm16418 GSM2545380 16
Gm16701 GSM2545380 181
Aldh9a1 GSM2545380 1770
Mgst3 GSM2545380 2151
Lrrc52 GSM2545380 5
Rxrg GSM2545380 49
Lmx1a GSM2545380 72
Pbx1 GSM2545380 4795We now have 20 rows in our combined dataset, and the same number of
columns. Note that concat is a function of the
pd module, as opposed to a dataframe method. It takes in a
list of dataframes, and outputs a combined dataframe.
If one dataframe has columns which don’t exist in the other, these
values are filled in with NaN.
PYTHON
rnaseq_mini_time = rnaseq_df.loc[:,["sample", "expression","time"]].iloc[10:20,:]
print(rnaseq_mini_time)OUTPUT
sample expression time
gene
Tnc GSM2545336 219 8
Trf GSM2545336 9719 8
Tubb2b GSM2545336 2245 8
Fads1 GSM2545336 6498 8
Lxn GSM2545336 1744 8
Prr18 GSM2545336 1284 8
Cmtm5 GSM2545336 1381 8
Enpp1 GSM2545336 388 8
Clic4 GSM2545336 5795 8
Tm6sf2 GSM2545336 32 8PYTHON
mini_dfs = [rnaseq_mini, rnaseq_mini_time, rnaseq_mini_tail]
combined_df = pd.concat(mini_dfs)
print(combined_df)OUTPUT
sample expression time
gene
Asl GSM2545336 1170 NaN
Apod GSM2545336 36194 NaN
Cyp2d22 GSM2545336 4060 NaN
Klk6 GSM2545336 287 NaN
Fcrls GSM2545336 85 NaN
Slc2a4 GSM2545336 782 NaN
Exd2 GSM2545336 1619 NaN
Gjc2 GSM2545336 288 NaN
Plp1 GSM2545336 43217 NaN
Gnb4 GSM2545336 1071 NaN
Tnc GSM2545336 219 8.0
Trf GSM2545336 9719 8.0
Tubb2b GSM2545336 2245 8.0
Fads1 GSM2545336 6498 8.0
Lxn GSM2545336 1744 8.0
Prr18 GSM2545336 1284 8.0
Cmtm5 GSM2545336 1381 8.0
Enpp1 GSM2545336 388 8.0
Clic4 GSM2545336 5795 8.0
Tm6sf2 GSM2545336 32 8.0
Dusp27 GSM2545380 15 NaN
Mael GSM2545380 4 NaN
Gm16418 GSM2545380 16 NaN
Gm16701 GSM2545380 181 NaN
Aldh9a1 GSM2545380 1770 NaN
Mgst3 GSM2545380 2151 NaN
Lrrc52 GSM2545380 5 NaN
Rxrg GSM2545380 49 NaN
Lmx1a GSM2545380 72 NaN
Pbx1 GSM2545380 4795 NaNMerge or join data frames to add additional columns.
As opposed to concatenating data, we instead might want to add additional columns to a dataframe. We’ve already seen how to add a new column based on a calculation, but often we have some other data table we want to combine.
If we know that the rows are in the same order, we can use the same syntax we use to assign new columns. However, this is often not the case.
PYTHON
# There is an ongoing idelogical debate among developers whether variables like this should be named:
# url1, url2, and url3
# url0, url1, and url2 or
# url, url2, and url3
url1 = "https://raw.githubusercontent.com/ccb-hms/workbench-python-workshop/main/episodes/data/annot1.csv"
url2 = "https://raw.githubusercontent.com/ccb-hms/workbench-python-workshop/main/episodes/data/annot2.csv"
url3 = "https://raw.githubusercontent.com/ccb-hms/workbench-python-workshop/main/episodes/data/annot3.csv"
# Here .sample is being used to shuffle the dataframe rows into a random order
annot1 = pd.read_csv(url1, index_col=0).sample(frac=1)
annot2 = pd.read_csv(url2, index_col=0).sample(frac=1)
annot3 = pd.read_csv(url3, index_col=0).sample(frac=1)
print(annot1)OUTPUT
gene_description
gene
Fcrls Fc receptor-like S, scavenger receptor [Source...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Klk6 kallikrein related-peptidase 6 [Source:MGI Sym...
Gnb4 guanine nucleotide binding protein (G protein)...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...OUTPUT
sample expression \
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Klk6 GSM2545336 287
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
gene_description
gene
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Klk6 kallikrein related-peptidase 6 [Source:MGI Sym...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Gnb4 guanine nucleotide binding protein (G protein)... We have combined the two dataframes to add the
gene_description column. By default, join
looks at the index column of the left and right dataframe, combining
rows when it finds matches. The data used to determine which rows should
be combined between the dataframes is referred to as what is being
joined on, or the keys. Here, we would say we are joining on
the index columns, or the index columns are the keys. The row order and
column order depends on which dataframe is on the left.
OUTPUT
gene_description sample \
gene
Fcrls Fc receptor-like S, scavenger receptor [Source... GSM2545336
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym... GSM2545336
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88... GSM2545336
Klk6 kallikrein related-peptidase 6 [Source:MGI Sym... GSM2545336
Gnb4 guanine nucleotide binding protein (G protein)... GSM2545336
Cyp2d22 cytochrome P450, family 2, subfamily d, polype... GSM2545336
Slc2a4 solute carrier family 2 (facilitated glucose t... GSM2545336
Asl argininosuccinate lyase [Source:MGI Symbol;Acc... GSM2545336
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb... GSM2545336
Exd2 exonuclease 3'-5' domain containing 2 [Source:... GSM2545336
expression
gene
Fcrls 85
Plp1 43217
Apod 36194
Klk6 287
Gnb4 1071
Cyp2d22 4060
Slc2a4 782
Asl 1170
Gjc2 288
Exd2 1619
And the index columns do not need to have the same name.
OUTPUT
description
external_gene_name
Slc2a4 solute carrier family 2 (facilitated glucose t...
Gnb4 guanine nucleotide binding protein (G protein)...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Klk6 kallikrein related-peptidase 6 [Source:MGI Sym...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...OUTPUT
sample expression \
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Klk6 GSM2545336 287
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
description
gene
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Klk6 kallikrein related-peptidase 6 [Source:MGI Sym...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Gnb4 guanine nucleotide binding protein (G protein)... join by default uses the indices of the dataframes it
combines. To change this, we can use the on parameter. For
instance, let’s say we want to combine our sample metadata back with
rnaseq_mini.
PYTHON
url = "https://raw.githubusercontent.com/ccb-hms/workbench-python-workshop/main/episodes/data/metadata.csv"
metadata = pd.read_csv(url, index_col=0)
print(metadata)OUTPUT
organism age sex infection strain time tissue \
sample
GSM2545336 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum
GSM2545337 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum
GSM2545338 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum
GSM2545339 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum
GSM2545340 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum
GSM2545341 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum
GSM2545342 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum
GSM2545343 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum
GSM2545344 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum
GSM2545345 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum
GSM2545346 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum
GSM2545347 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum
GSM2545348 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum
GSM2545349 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum
GSM2545350 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum
GSM2545351 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum
GSM2545352 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum
GSM2545353 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum
GSM2545354 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum
GSM2545362 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum
GSM2545363 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum
GSM2545380 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum
mouse
sample
GSM2545336 14
GSM2545337 9
GSM2545338 10
GSM2545339 15
GSM2545340 18
GSM2545341 6
GSM2545342 5
GSM2545343 11
GSM2545344 22
GSM2545345 13
GSM2545346 23
GSM2545347 24
GSM2545348 8
GSM2545349 7
GSM2545350 1
GSM2545351 16
GSM2545352 21
GSM2545353 4
GSM2545354 2
GSM2545362 20
GSM2545363 12
GSM2545380 19 OUTPUT
sample expression organism age sex infection \
gene
Asl GSM2545336 1170 Mus musculus 8 Female InfluenzaA
Apod GSM2545336 36194 Mus musculus 8 Female InfluenzaA
Cyp2d22 GSM2545336 4060 Mus musculus 8 Female InfluenzaA
Klk6 GSM2545336 287 Mus musculus 8 Female InfluenzaA
Fcrls GSM2545336 85 Mus musculus 8 Female InfluenzaA
Slc2a4 GSM2545336 782 Mus musculus 8 Female InfluenzaA
Exd2 GSM2545336 1619 Mus musculus 8 Female InfluenzaA
Gjc2 GSM2545336 288 Mus musculus 8 Female InfluenzaA
Plp1 GSM2545336 43217 Mus musculus 8 Female InfluenzaA
Gnb4 GSM2545336 1071 Mus musculus 8 Female InfluenzaA
strain time tissue mouse
gene
Asl C57BL/6 8 Cerebellum 14
Apod C57BL/6 8 Cerebellum 14
Cyp2d22 C57BL/6 8 Cerebellum 14
Klk6 C57BL/6 8 Cerebellum 14
Fcrls C57BL/6 8 Cerebellum 14
Slc2a4 C57BL/6 8 Cerebellum 14
Exd2 C57BL/6 8 Cerebellum 14
Gjc2 C57BL/6 8 Cerebellum 14
Plp1 C57BL/6 8 Cerebellum 14
Gnb4 C57BL/6 8 Cerebellum 14 Note that if we want to join on columns between dataframes which have different names we can simply rename the columns we wish to join on. If there is a reason we don’t want to do this, we can instead use the more powerful but harder to use pandas merge function.
Missing and duplicate data
In the above join, there were a few other things going on. The first
is that multiple rows of rnaseq_mini contain the same
sample. Pandas by default will repeat the rows wherever needed when
joining.
The second is that the metadata dataframe contains
samples which are not present in rnaseq_mini. By default,
they were dropped from the dataframe. However, there might be cases
where we want to keep our data. Let’s explore how joining handles
missing data with another example:
OUTPUT
gene_description
gene
mt-Rnr1 mitochondrially encoded 12S rRNA [Source:MGI S...
mt-Rnr2 mitochondrially encoded 16S rRNA [Source:MGI S...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gnb4 guanine nucleotide binding protein (G protein)...
mt-Tl1 mitochondrially encoded tRNA leucine 1 [Source...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
mt-Tv mitochondrially encoded tRNA valine [Source:MG...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
mt-Tf mitochondrially encoded tRNA phenylalanine [So...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...Missing data in joins
What data is missing between annot3 and
rnaseq_mini?
Try joining them. How is this missing data handled?
There are both genes in annot3 but not in
rnaseq_mini, and genes in rnaseq_mini not in
annot3.
When we join, we keep rows from rnaseq_mini with missing data and
drop rows from annot3 with missing data.
OUTPUT
sample expression \
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Klk6 GSM2545336 287
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
gene_description
gene
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Klk6 NaN
Fcrls Fc receptor-like S, scavenger receptor [Source...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Gnb4 guanine nucleotide binding protein (G protein)... Types of joins
The reason we see the above behavior is because by default pandas performs a left join.
We can change the type of join performed by changing the
how argument of the join method.
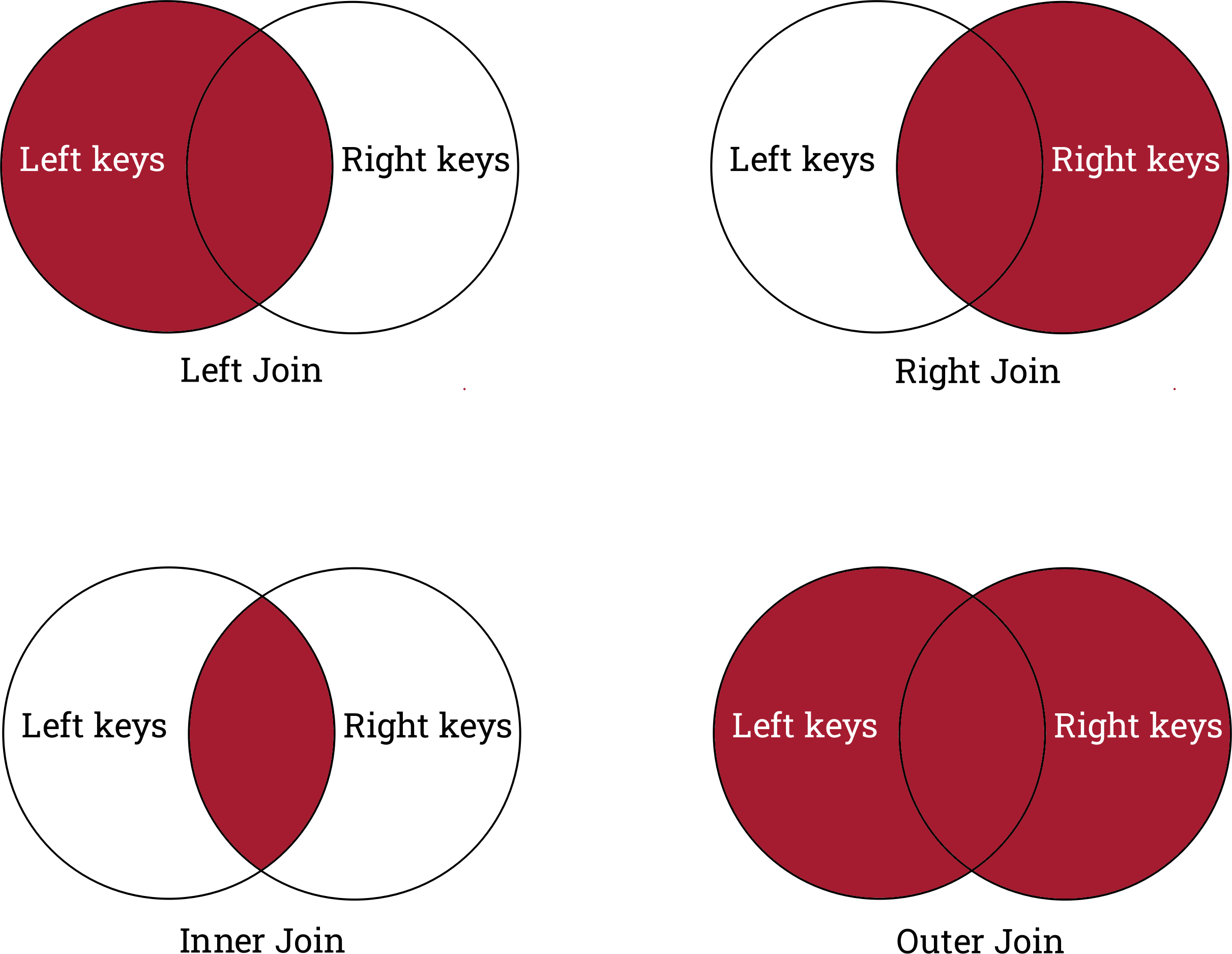
A right join saves all genes in the right dataframe,
but drops and genes unique to rnaseq_mini.
OUTPUT
sample expression \
gene
Plp1 GSM2545336 43217.0
Exd2 GSM2545336 1619.0
Gjc2 GSM2545336 288.0
mt-Rnr2 NaN NaN
Gnb4 GSM2545336 1071.0
mt-Rnr1 NaN NaN
Apod GSM2545336 36194.0
Fcrls GSM2545336 85.0
mt-Tv NaN NaN
mt-Tf NaN NaN
Slc2a4 GSM2545336 782.0
Cyp2d22 GSM2545336 4060.0
mt-Tl1 NaN NaN
Asl GSM2545336 1170.0
gene_description
gene
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
mt-Rnr2 mitochondrially encoded 16S rRNA [Source:MGI S...
Gnb4 guanine nucleotide binding protein (G protein)...
mt-Rnr1 mitochondrially encoded 12S rRNA [Source:MGI S...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Fcrls Fc receptor-like S, scavenger receptor [Source...
mt-Tv mitochondrially encoded tRNA valine [Source:MG...
mt-Tf mitochondrially encoded tRNA phenylalanine [So...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
mt-Tl1 mitochondrially encoded tRNA leucine 1 [Source...
Asl argininosuccinate lyase [Source:MGI Symbol;Acc... An inner join only keeps genes shared by the dataframes, and drops all genes which are only in one dataframe.
OUTPUT
sample expression \
gene
Asl GSM2545336 1170
Apod GSM2545336 36194
Cyp2d22 GSM2545336 4060
Fcrls GSM2545336 85
Slc2a4 GSM2545336 782
Exd2 GSM2545336 1619
Gjc2 GSM2545336 288
Plp1 GSM2545336 43217
Gnb4 GSM2545336 1071
gene_description
gene
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Slc2a4 solute carrier family 2 (facilitated glucose t...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Gnb4 guanine nucleotide binding protein (G protein)... Finally, an outer join keeps all genes across both dataframes.
OUTPUT
sample expression \
gene
Apod GSM2545336 36194.0
Asl GSM2545336 1170.0
Cyp2d22 GSM2545336 4060.0
Exd2 GSM2545336 1619.0
Fcrls GSM2545336 85.0
Gjc2 GSM2545336 288.0
Gnb4 GSM2545336 1071.0
Klk6 GSM2545336 287.0
Plp1 GSM2545336 43217.0
Slc2a4 GSM2545336 782.0
mt-Rnr1 NaN NaN
mt-Rnr2 NaN NaN
mt-Tf NaN NaN
mt-Tl1 NaN NaN
mt-Tv NaN NaN
gene_description
gene
Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88...
Asl argininosuccinate lyase [Source:MGI Symbol;Acc...
Cyp2d22 cytochrome P450, family 2, subfamily d, polype...
Exd2 exonuclease 3'-5' domain containing 2 [Source:...
Fcrls Fc receptor-like S, scavenger receptor [Source...
Gjc2 gap junction protein, gamma 2 [Source:MGI Symb...
Gnb4 guanine nucleotide binding protein (G protein)...
Klk6 NaN
Plp1 proteolipid protein (myelin) 1 [Source:MGI Sym...
Slc2a4 solute carrier family 2 (facilitated glucose t...
mt-Rnr1 mitochondrially encoded 12S rRNA [Source:MGI S...
mt-Rnr2 mitochondrially encoded 16S rRNA [Source:MGI S...
mt-Tf mitochondrially encoded tRNA phenylalanine [So...
mt-Tl1 mitochondrially encoded tRNA leucine 1 [Source...
mt-Tv mitochondrially encoded tRNA valine [Source:MG... Duplicate column names
One common challenge we encounter when combining datasets from different sources is that they have identical column names.
Imagine that you’ve collected a second set of observations from
samples which are stored in a identically structured file. We can
simulate this by generating some random numbers in a copy of
rnaseq_mini.
PYTHON
new_mini = rnaseq_mini.copy()
new_mini["expression"] = pd.Series(range(50,50000)).sample(int(10), replace=True).array
print(new_mini)Note: as these are pseudo-random numbers your exact values will be different
OUTPUT
sample expression
gene
Asl GSM2545336 48562
Apod GSM2545336 583
Cyp2d22 GSM2545336 39884
Klk6 GSM2545336 6161
Fcrls GSM2545336 10318
Slc2a4 GSM2545336 15991
Exd2 GSM2545336 44471
Gjc2 GSM2545336 40629
Plp1 GSM2545336 23146
Gnb4 GSM2545336 22506Try joining rnaseq_mini and new_mini. What
happens? Take a look at the lsuffix and
rsuffix arguments for join. How can you use
these to improve your joined dataframe?
When we try to join these dataframes we get an error.
ERROR
---------------------------------------------------------------------------
ValueError Traceback (most recent call last)
Cell In[17], line 1
----> 1 rnaseq_mini.join(new_mini)
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\frame.py:9976, in DataFrame.join(self, other, on, how, lsuffix, rsuffix, sort, validate)
9813 def join(
9814 self,
9815 other: DataFrame | Series | list[DataFrame | Series],
(...)
9821 validate: str | None = None,
9822 ) -> DataFrame:
9823 """
9824 Join columns of another DataFrame.
9825
(...)
9974 5 K1 A5 B1
9975 """
-> 9976 return self._join_compat(
9977 other,
9978 on=on,
9979 how=how,
9980 lsuffix=lsuffix,
9981 rsuffix=rsuffix,
9982 sort=sort,
9983 validate=validate,
9984 )
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\frame.py:10015, in DataFrame._join_compat(self, other, on, how, lsuffix, rsuffix, sort, validate)
10005 if how == "cross":
10006 return merge(
10007 self,
10008 other,
(...)
10013 validate=validate,
10014 )
> 10015 return merge(
10016 self,
10017 other,
10018 left_on=on,
10019 how=how,
10020 left_index=on is None,
10021 right_index=True,
10022 suffixes=(lsuffix, rsuffix),
10023 sort=sort,
10024 validate=validate,
10025 )
10026 else:
10027 if on is not None:
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\reshape\merge.py:124, in merge(left, right, how, on, left_on, right_on, left_index, right_index, sort, suffixes, copy, indicator, validate)
93 @Substitution("\nleft : DataFrame or named Series")
94 @Appender(_merge_doc, indents=0)
95 def merge(
(...)
108 validate: str | None = None,
109 ) -> DataFrame:
110 op = _MergeOperation(
111 left,
112 right,
(...)
122 validate=validate,
123 )
--> 124 return op.get_result(copy=copy)
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\reshape\merge.py:775, in _MergeOperation.get_result(self, copy)
771 self.left, self.right = self._indicator_pre_merge(self.left, self.right)
773 join_index, left_indexer, right_indexer = self._get_join_info()
--> 775 result = self._reindex_and_concat(
776 join_index, left_indexer, right_indexer, copy=copy
777 )
778 result = result.__finalize__(self, method=self._merge_type)
780 if self.indicator:
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\reshape\merge.py:729, in _MergeOperation._reindex_and_concat(self, join_index, left_indexer, right_indexer, copy)
726 left = self.left[:]
727 right = self.right[:]
--> 729 llabels, rlabels = _items_overlap_with_suffix(
730 self.left._info_axis, self.right._info_axis, self.suffixes
731 )
733 if left_indexer is not None:
734 # Pinning the index here (and in the right code just below) is not
735 # necessary, but makes the `.take` more performant if we have e.g.
736 # a MultiIndex for left.index.
737 lmgr = left._mgr.reindex_indexer(
738 join_index,
739 left_indexer,
(...)
744 use_na_proxy=True,
745 )
File ~\anaconda3\envs\ml-env\lib\site-packages\pandas\core\reshape\merge.py:2458, in _items_overlap_with_suffix(left, right, suffixes)
2455 lsuffix, rsuffix = suffixes
2457 if not lsuffix and not rsuffix:
-> 2458 raise ValueError(f"columns overlap but no suffix specified: {to_rename}")
2460 def renamer(x, suffix):
2461 """
2462 Rename the left and right indices.
2463
(...)
2474 x : renamed column name
2475 """
ValueError: columns overlap but no suffix specified: Index(['sample', 'expression'], dtype='object')We need to give the datasets suffixes so that there is no column name collision.
OUTPUT
sample_exp1 expression_exp1 sample_exp2 expression_exp2
gene
Asl GSM2545336 1170 GSM2545336 29016
Apod GSM2545336 36194 GSM2545336 46560
Cyp2d22 GSM2545336 4060 GSM2545336 1823
Klk6 GSM2545336 287 GSM2545336 27428
Fcrls GSM2545336 85 GSM2545336 45369
Slc2a4 GSM2545336 782 GSM2545336 31129
Exd2 GSM2545336 1619 GSM2545336 21478
Gjc2 GSM2545336 288 GSM2545336 34747
Plp1 GSM2545336 43217 GSM2545336 46074
Gnb4 GSM2545336 1071 GSM2545336 16370While this works, we now have duplicate sample
columns.
To avoid this, we could either drop the sample column in
one of the dataframes before joining, or use merge to join
on multiple columns and reset the index afterward.
PYTHON
#Option 1: Drop the column
print(rnaseq_mini.join(new_mini.drop('sample',axis=1), lsuffix="_exp1", rsuffix="_exp2"))OUTPUT
sample expression_exp1 expression_exp2
gene
Asl GSM2545336 1170 29016
Apod GSM2545336 36194 46560
Cyp2d22 GSM2545336 4060 1823
Klk6 GSM2545336 287 27428
Fcrls GSM2545336 85 45369
Slc2a4 GSM2545336 782 31129
Exd2 GSM2545336 1619 21478
Gjc2 GSM2545336 288 34747
Plp1 GSM2545336 43217 46074
Gnb4 GSM2545336 1071 16370PYTHON
#Option 2: Merging on gene and sample
print(pd.merge(rnaseq_mini, new_mini, on=["gene","sample"]))OUTPUT
sample expression_x expression_y
gene
Asl GSM2545336 1170 29016
Apod GSM2545336 36194 46560
Cyp2d22 GSM2545336 4060 1823
Klk6 GSM2545336 287 27428
Fcrls GSM2545336 85 45369
Slc2a4 GSM2545336 782 31129
Exd2 GSM2545336 1619 21478
Gjc2 GSM2545336 288 34747
Plp1 GSM2545336 43217 46074
Gnb4 GSM2545336 1071 16370Note that pd.merge does not throw an error when dealing
with duplicate column names, but instead automatically uses the suffixes
_x and _y. We could change these defaults with
the suffixes argument.